 Professor
Professor
Research Areas
Transcription Regulation, Oncogenic Signaling, Metabolism and Anti-tumor Immune Regulation
Research interests and long-term vision
The regulation of gene expression and signal transduction pathways play a fundamental role in cellular survival and adaptability across all living organisms, from single-celled bacteria to complex mammals. In cancer, these regulatory mechanisms often become hijacked, resulting in uncontrolled growth and metastasis. My laboratory is dedicated to dissecting these regulatory systems to uncover cancer-specific vulnerabilities that can be targeted for more precise and effective treatments. Our approach integrates both unbiased, functional genomics techniques and hypothesis-driven research to advance our understanding of cancer. The primary objective of my research program is to identify and characterize critical cancer-specific pathways and regulatory mechanisms, ultimately contributing to novel therapeutic options. Below, I outline three primary areas of my lab’s research focus and discuss our ongoing projects and future directions.
Research Direction 1: Mechanisms of oncogene-driven Transcriptional silencing of tumor suppressor genes
Background
Oncogenic mutations or gene amplification are two common mechanisms of activation of oncogenic pathways. One of the primary mechanisms through which mutant oncogenes promote cellular transformation and cancer growth is by orchestrating the silencing of tumor suppressor genes (TSGs). This silencing occurs via two fundamental processes: promoter DNA hypermethylation and transcriptionally repressive histone modifications. Notably, certain prominent TSGs, including CDKN2A and RASSF1A, are predominantly inactivated through DNA methylation-based transcriptional gene silencing, as opposed to genetic mechanisms such as mutation or deletion. Perturbations in epigenetic silencing directly contribute to the initiation and progression of multiple diseases, including cancer. We have worked extensively to identify mechanisms by which oncogenes drive transcriptional silencing of tumor suppressor genes (TSGs) in cancer cells. These studies have identified multiple new pathways and mechanisms through which oncogenes drive transcriptional silencing of TSGs.
Research accomplishments
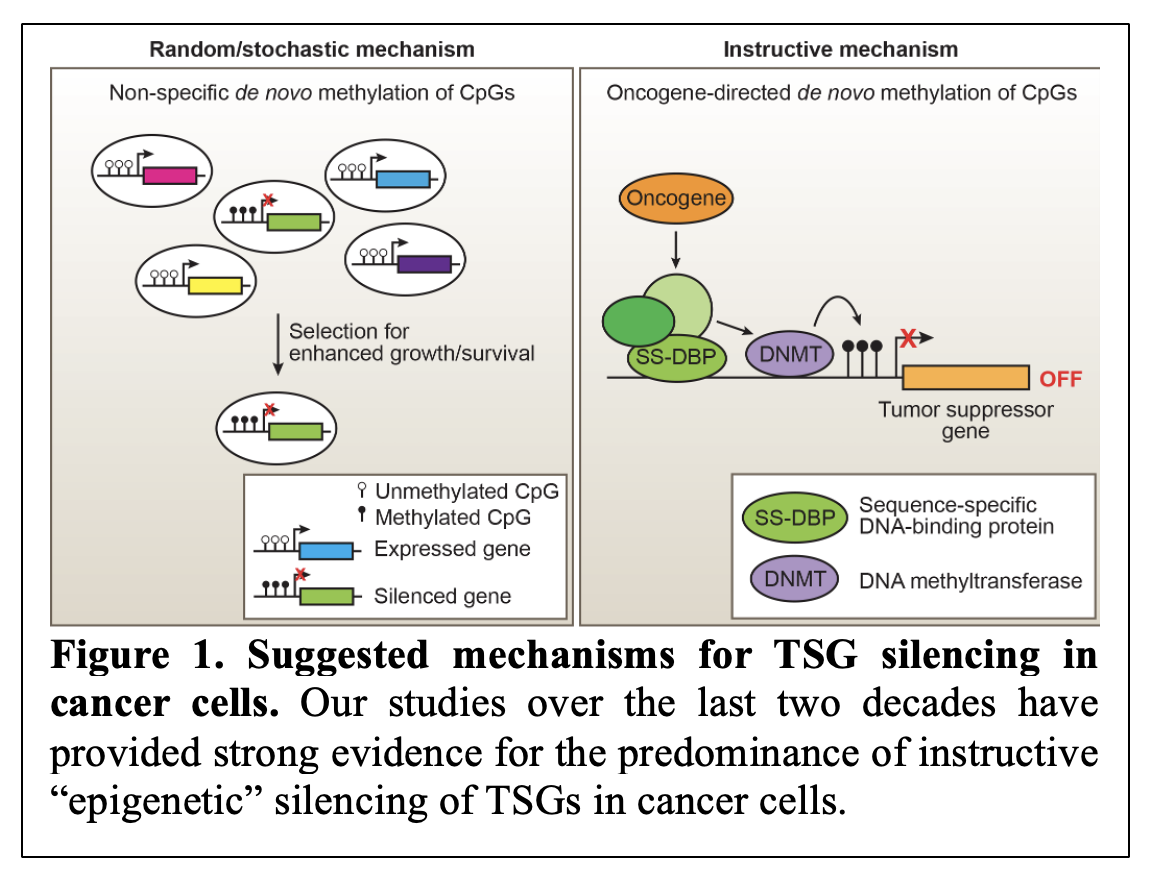 Over the last two decades, we have worked toward understanding the mechanism behind transcriptional silencing of tumor suppressor genes (TSGs). When we began our work, there were two prevailing models of how TSGs can be transcriptionally silenced in cancer cells. These models included stochastic model of TSG silencing and an alternative model describing a more controlled process of TSG silencing referred to as instructive gene silencing (Figure 1). Over the years, our research has uncovered significant evidence in support of the instructive model (Gazin/Wajapeyee et al., Nature, 2007; Palakurthy et al., Molecular Cell, 2009; Wajapeyee et al., Genes and Development, 2013; Forloni et al., Cell Reports 2016; Chava et al., iScience, 2022) 1-6.
Over the last two decades, we have worked toward understanding the mechanism behind transcriptional silencing of tumor suppressor genes (TSGs). When we began our work, there were two prevailing models of how TSGs can be transcriptionally silenced in cancer cells. These models included stochastic model of TSG silencing and an alternative model describing a more controlled process of TSG silencing referred to as instructive gene silencing (Figure 1). Over the years, our research has uncovered significant evidence in support of the instructive model (Gazin/Wajapeyee et al., Nature, 2007; Palakurthy et al., Molecular Cell, 2009; Wajapeyee et al., Genes and Development, 2013; Forloni et al., Cell Reports 2016; Chava et al., iScience, 2022) 1-6.
These studies have documented that epigenetic silencing of TSGs in cancer cells is a non-random, highly orchestrated process controlled by specific oncogenes through sequential recruitment of transcriptional repressors to TSG promoters. Notably, our studies also revealed that the TSG transcriptional silencing was not truly epigenetic, as it required continuous activity of oncogenic pathways and could be rapidly reversed by the suppression of oncogenic pathways using small molecule inhibitors, such as MEK and PI3K inhibitors in the context of oncogenic RAS proteins (reviewed by Kevin Strhul in eLife, 2014) 7.
Future research direction
Building on these findings, my lab aims to identify additional pathways and factors that contribute to TSG silencing across different cancer types. We are employing large-scale functional genomics screens to discover novel regulators involved in TSG transcriptional silencing, with a particular focus on factors that may be manipulated therapeutically. By expanding our studies to additional oncogenes and cancer models, we anticipate developing a comprehensive map of instructive gene silencing, paving the way for targeted epigenetic therapies.
Furthermore, using genetic screening approaches such as those described above, we are identifying new factors that are necessary for TSG silencing and gene imprinting in a variety of cancer types. A related area of research is to compare different mutants of the same oncogenes and assess their ability to cause epigenetic TSG silencing. For example, our collective body of work, encompassing both published and unpublished studies, provides preliminary evidence suggesting that oncogenic KRAS and EGFR mutations exhibit distinct, tissue-specific, and cancer-relevant effects by differentially regulating gene expression. In the context of oncogenic epidermal growth factor receptor (EGFR) mutations, we find that, in addition to activating an instructive pathway to cause TSG silencing, there is a significant level of stochastic gene silencing that is required for the maintenance of a specific oncogenic EGFR mutant in cancer cells. Our future studies will also aim to understand the mechanisms underlying this regulation and to investigate transcriptional heterogeneity in the context of specific signaling pathways, for example in the context of the RAS/RAF and EGFR pathways.
Research Direction 2: Identification of Drivers of Cancer Initiation and Progression
Background
Cancer is a complex disease and its initiation and progression are driven by multiple factors and pathways. Previous discoveries of cancer drivers such as RAS, BRAF and EGFR have paved the way for mutant-specific drug development and their translation for cancer treatment to the clinic. These previous successes highlight the importance of discovering additional drivers of clinical value for cancer treatment. Using various cancer models, particularly those for which there are limited clinical opportunities, we have performed studies to identify previously undocumented drivers of cancer initiation and progression with the goal of developing new therapies.
Research Accomplishments
For identifying the drivers of tumor initiation and progression, we have used both genetic screens, genomics/transcriptomics approaches, and hypothesis-driven projects. For example, in collaboration with Dr. Michael R. Green, our lab identified TRIM 37 as a novel histone H2A ubiquitin ligase and breast cancer oncogene (Bhatnagar et al., Nature, 2014) 1 and also identified a number of novel lung cancer tumor suppressors (Lin et al., Cancer Discovery, 2014) 8. Additionally, using more directed cancer patient database analysis-based experiments, we have identified transcription factor KLF7 and HOXC6 as important transcriptional drivers of pancreatic ductal adenocarcinoma tumor growth and metastasis (Gupta et al., PNAS, 2020; Malvi et al., Cell Reports Medicine, 2023) 9, 10.
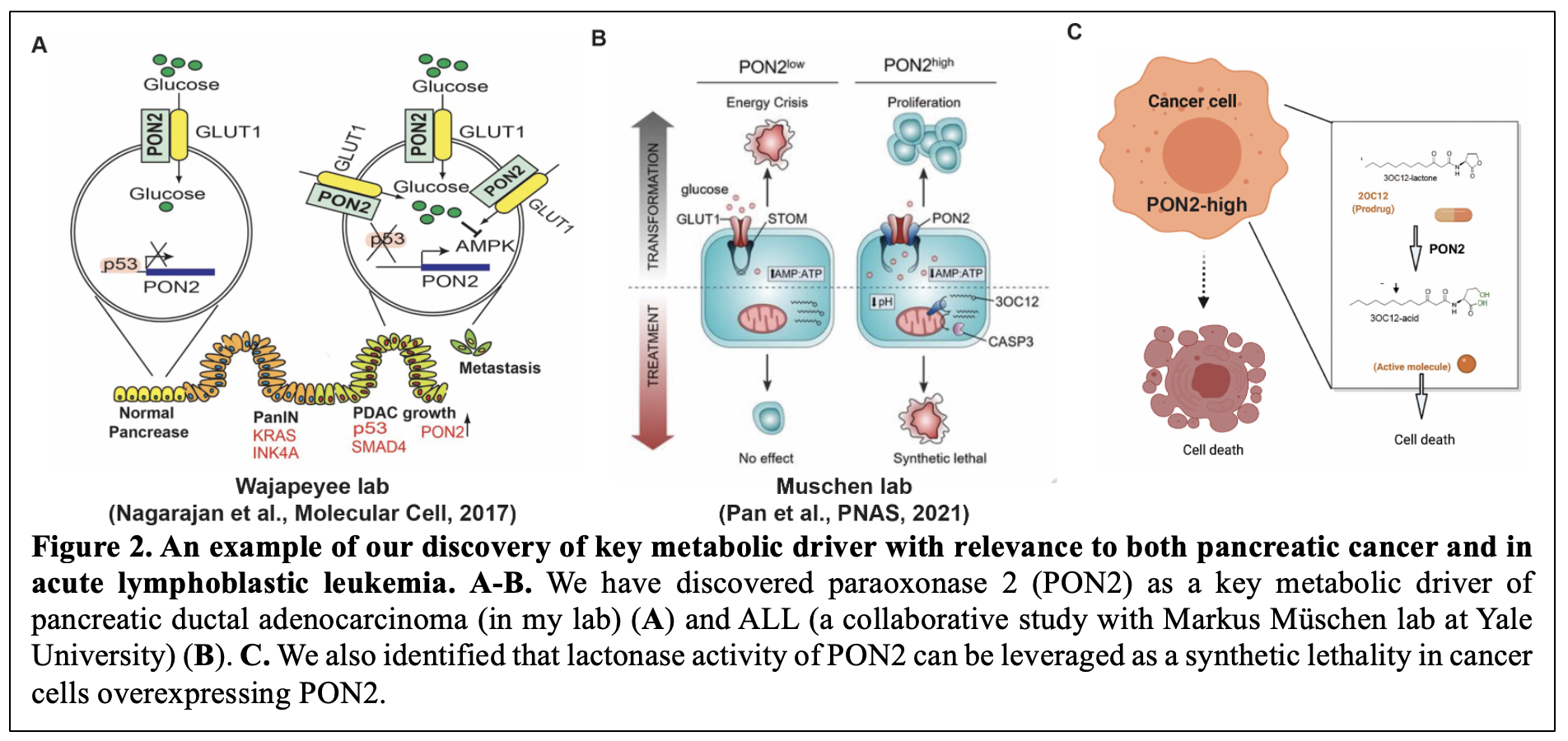 Furthermore, to identify cancer type-specific metabolic vulnerabilities, we integrated transcriptional profiles of cancer mRNA expression patterns with functional genomics. Using this approach, we uncovered multiple metabolic pathways essential for cancer cell survival (Gupta et al., Oncogene, 2017; Nagarajan et al., Molecular Cell, 2017; Rawat et al., Oncogene, 2021) 11-13. In particular, we discovered Paraoxonase-2 (PON2) as a key metabolic driver of pancreatic ductal adenocarcinoma (in my lab) and acute lymphoblastic leukemia (ALL) (a collaborative study with Marcus München’s lab at Yale University) (Figure 2A-2B). These studies also identified that lactonase activity of PON2 can be leveraged as a synthetic lethality using a pro-drug approach to specifically eradicate PON2 overexpressing tumors (Figure 2C) 13, 14. Further studies are identifying new roles and mechanisms of action for PON2 in cancer.
Furthermore, to identify cancer type-specific metabolic vulnerabilities, we integrated transcriptional profiles of cancer mRNA expression patterns with functional genomics. Using this approach, we uncovered multiple metabolic pathways essential for cancer cell survival (Gupta et al., Oncogene, 2017; Nagarajan et al., Molecular Cell, 2017; Rawat et al., Oncogene, 2021) 11-13. In particular, we discovered Paraoxonase-2 (PON2) as a key metabolic driver of pancreatic ductal adenocarcinoma (in my lab) and acute lymphoblastic leukemia (ALL) (a collaborative study with Marcus München’s lab at Yale University) (Figure 2A-2B). These studies also identified that lactonase activity of PON2 can be leveraged as a synthetic lethality using a pro-drug approach to specifically eradicate PON2 overexpressing tumors (Figure 2C) 13, 14. Further studies are identifying new roles and mechanisms of action for PON2 in cancer.
Future research direction
Based on our previous success in the context of identifying new drug targets both in naïve cancers and drug-resistant tumors, we will continue our efforts in these directions. In particular, we are expanding our studies in relation to metabolic adaptation, specially its effect on transcriptional deregulation in cancer cells. Similarly, we are studying mechanisms of adaptation to metabolic pathway inhibition and their impact on tumor growth and metastasis progression. These efforts are expected to continue in the future. Finally, related to cancer driver discoveries, we are also starting to take a more careful look at factors that affect one step versus another in tumor growth or metastasis. For example, ongoing studies in our lab are now identifying and characterizing new metastasis drivers and performing mechanistic studies to understand their mode of action.
Research Direction 3: Immune-mediated Cancer Control
Background
The immune system plays a central role in suppressing tumor initiation and progression, and consists of the adaptive and innate immune systems. The adaptive immune system includes lymphocytes such as B cells and T cells, each with distinct roles in either suppressing or promoting cancer. The innate immune system provides the first line of defense against cancer through rapid and non-specific responses, involving various cells like neutrophils, macrophages, dendritic cells, NK cells, eosinophils, basophils, mast cells, monocytes, and NKT cells. These contribute uniquely to either detecting and destroying cancer cells or enabling tumor progression. Cancer cells survive the cytotoxic effects of both the adaptive and innate immune systems, a process called immune evasion. In recent years, we have focused our efforts on transcriptional pathways that control immune evasion with a particular focus on NK cells and some other poorly studied immune cell types. The goal of these studies is to identify alternative approaches to increase anti-tumor immunity against cancer cells.
Research accomplishments
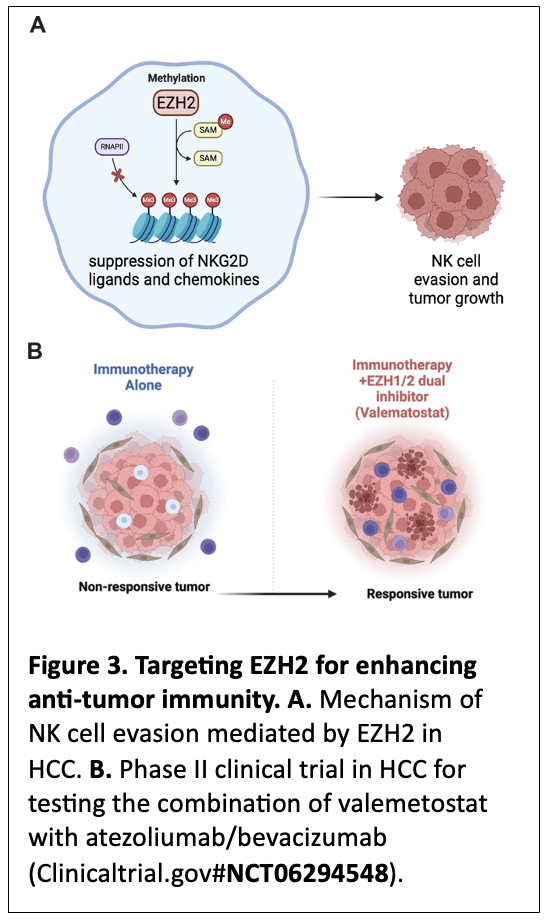 A key area of our research focuses on understanding how the innate immune system, specifically natural killer (NK) cells, can be leveraged for cancer therapy. We recently demonstrated that inhibition of EZH2, a protein in the polycomb repressive complex 2 (PRC2), enhances NK cell-mediated killing of hepatocellular carcinoma (HCC) cells (Bugide et al., PNAS, 2018; Bugide et al., PNAS, 2021) 15, 16 (Figure 3A). In particular, we have discovered that EZH2 suppresses the expression of NKG2D ligands on HCC cells and prevent their eradication by NK cells 15. Additionally, we have identified that EZH2 can suppress NK cell recruitment to tumor microenvironment by suppressing the expression of chemokines, such as CXCL10 16. Our observations in HCC are recapitulated by Marcus Ruscetti’s lab at UMass Chan Medical School in pancreatic cancers 17. These findings have significant implications for cancer immunotherapy, because they demonstrate that epigenetic modulation, such as suppression of EZH2, enhances NK cell-mediated anti-tumor immunity. Based on these preclinical studies from our lab and those of others that identified EZH2 as an immunosuppressive protein, we have initiated a Phase II clinical trial supported by Daiichi Sankyo in collaboration with a Medical Oncology colleague here at the University of Alabama at Birmingham. The goal of this Phase II trial is to establish that co-targeting of EZH2/1 using Valemetostat in combination with front-line HCC therapies such as Atezolizumab and Bevacizumab can result in significantly better and more durable responses in HCC patients. Based on our preclinical studies, we rationalize that this approach in the clinic may even allow tumor non-responsive or minimally responsive to front-line immunotherapies to become responsive by reversing the immunosuppressive state driven by EZH2 (Figure 3B). A successful outcome of this clinical trial will provide new and better therapeutic options to HCC patients, with implications for other cancer types, such as pancreatic cancer.
A key area of our research focuses on understanding how the innate immune system, specifically natural killer (NK) cells, can be leveraged for cancer therapy. We recently demonstrated that inhibition of EZH2, a protein in the polycomb repressive complex 2 (PRC2), enhances NK cell-mediated killing of hepatocellular carcinoma (HCC) cells (Bugide et al., PNAS, 2018; Bugide et al., PNAS, 2021) 15, 16 (Figure 3A). In particular, we have discovered that EZH2 suppresses the expression of NKG2D ligands on HCC cells and prevent their eradication by NK cells 15. Additionally, we have identified that EZH2 can suppress NK cell recruitment to tumor microenvironment by suppressing the expression of chemokines, such as CXCL10 16. Our observations in HCC are recapitulated by Marcus Ruscetti’s lab at UMass Chan Medical School in pancreatic cancers 17. These findings have significant implications for cancer immunotherapy, because they demonstrate that epigenetic modulation, such as suppression of EZH2, enhances NK cell-mediated anti-tumor immunity. Based on these preclinical studies from our lab and those of others that identified EZH2 as an immunosuppressive protein, we have initiated a Phase II clinical trial supported by Daiichi Sankyo in collaboration with a Medical Oncology colleague here at the University of Alabama at Birmingham. The goal of this Phase II trial is to establish that co-targeting of EZH2/1 using Valemetostat in combination with front-line HCC therapies such as Atezolizumab and Bevacizumab can result in significantly better and more durable responses in HCC patients. Based on our preclinical studies, we rationalize that this approach in the clinic may even allow tumor non-responsive or minimally responsive to front-line immunotherapies to become responsive by reversing the immunosuppressive state driven by EZH2 (Figure 3B). A successful outcome of this clinical trial will provide new and better therapeutic options to HCC patients, with implications for other cancer types, such as pancreatic cancer.
Similar to our finding in HCC, my laboratory in collaboration with Dr. Eddy Yang’s group (now at the University of Kentucky) has identified tumoral ring finger protein 2 (RNF2), a protein that is part of polycomb repressor complex 1, as a negative regulator of antitumor immunity in breast cancer. This study discovered that in syngeneic models of triple-negative breast cancer, deleting genes encoding the polycomb repressor complex 1 subunits such as RNF2, BMI1, or RSF1 genes led to durable tumor rejection and immune memory by enhancing the infiltration and activation of NK and CD4+ T cells. These results reveal an epigenetic reprogramming of the tumor-immune microenvironment that promotes lasting antitumor immunity (Zhang et al., Nature Cancer, 2021) 18. Collectively, the outcome of these findings identifies creative strategies to enhance anti-tumor immunity, focusing on the precise targeting of cancer cells.
Future Research Directions
In the future, we plan to continue our efforts to identify additional epigenetic and genetic factors that regulate NK cell activity against tumor cells, particularly in the context of solid tumors. In particular, we have recently expanded our studies to a series of difficult to treat cancers, such as uveal melanoma, a highly aggressive eye cancer with a strong propensity to metastasize to the liver. In uveal melanoma, we are in the process of identifying previously undocumented regulators of NK cells and other key immune cells of therapeutic value. Our efforts will include preclinical studies in immunocompetent humanized mouse models to better simulate the clinical scenario. Ultimately, we aim to develop clinical protocols to assess NK cell and other unique immune cell-based therapies in cancer patients.
Conclusion and Vision
The research directions outlined here highlight my lab’s commitment to advancing the field of cancer biology through a translational approach that bridges fundamental research and clinical applications. Through the integration of basic science discoveries with clinical applications, my goal is to develop therapies that address unmet needs in cancer treatment.
References
1. Bhatnagar S, Gazin C, Chamberlain L, Ou J, Zhu X, Tushir JS, Virbasius CM, Lin L, Zhu LJ, Wajapeyee N, Green MR. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature. 2014;516(7529):116-20. Epub 20141124. doi: 10.1038/nature13955. PubMed PMID: 25470042; PMCID: PMC4269325.
2. Chava S, Bugide S, Zhang X, Gupta R, Wajapeyee N. Betacellulin promotes tumor development and EGFR mutant lung cancer growth by stimulating the EGFR pathway and suppressing apoptosis. iScience. 2022;25(5):104211. Epub 20220406. doi: 10.1016/j.isci.2022.104211. PubMed PMID: 35494243; PMCID: PMC9048069.
3. Forloni M, Gupta R, Nagarajan A, Sun LS, Dong Y, Pirazzoli V, Toki M, Wurtz A, Melnick MA, Kobayashi S, Homer RJ, Rimm DL, Gettinger SJ, Politi K, Dogra SK, Wajapeyee N. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Rep. 2016;16(2):457-71. Epub 20160623. doi: 10.1016/j.celrep.2016.05.087. PubMed PMID: 27346347; PMCID: PMC4945411.
4. Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449(7165):1073-7. doi: 10.1038/nature06251. PubMed PMID: 17960246; PMCID: PMC2147719.
5. Palakurthy RK, Wajapeyee N, Santra MK, Gazin C, Lin L, Gobeil S, Green MR. Epigenetic silencing of the RASSF1A tumor suppressor gene through HOXB3-mediated induction of DNMT3B expression. Mol Cell. 2009;36(2):219-30. doi: 10.1016/j.molcel.2009.10.009. PubMed PMID: 19854132; PMCID: PMC2776651.
6. Wajapeyee N, Malonia SK, Palakurthy RK, Green MR. Oncogenic RAS directs silencing of tumor suppressor genes through ordered recruitment of transcriptional repressors. Genes Dev. 2013;27(20):2221-6. Epub 20131008. doi: 10.1101/gad.227413.113. PubMed PMID: 24105743; PMCID: PMC3814642.
7. Struhl K. Is DNA methylation of tumour suppressor genes epigenetic? Elife. 2014;3:e02475. Epub 20140312. doi: 10.7554/eLife.02475. PubMed PMID: 24623307; PMCID: PMC3949415.
8. Lin L, Chamberlain L, Pak ML, Nagarajan A, Gupta R, Zhu LJ, Wright CM, Fong KM, Wajapeyee N, Green MR. A large-scale RNAi-based mouse tumorigenesis screen identifies new lung cancer tumor suppressors that repress FGFR signaling. Cancer Discov. 2014;4(10):1168-81. Epub 20140711. doi: 10.1158/2159-8290.CD-13-0747. PubMed PMID: 25015643; PMCID: PMC4184919.
9. Gupta R, Malvi P, Parajuli KR, Janostiak R, Bugide S, Cai G, Zhu LJ, Green MR, Wajapeyee N. KLF7 promotes pancreatic cancer growth and metastasis by up-regulating ISG expression and maintaining Golgi complex integrity. Proc Natl Acad Sci U S A. 2020;117(22):12341-51. Epub 20200519. doi: 10.1073/pnas.2005156117. PubMed PMID: 32430335; PMCID: PMC7275752.
10. Malvi P, Chava S, Cai G, Hu K, Zhu LJ, Edwards YJK, Green MR, Gupta R, Wajapeyee N. HOXC6 drives a therapeutically targetable pancreatic cancer growth and metastasis pathway by regulating MSK1 and PPP2R2B. Cell Rep Med. 2023;4(11):101285. Epub 20231110. doi: 10.1016/j.xcrm.2023.101285. PubMed PMID: 37951219; PMCID: PMC10694669.
11. Gupta R, Yang Q, Dogra SK, Wajapeyee N. Serine hydroxymethyl transferase 1 stimulates pro-oncogenic cytokine expression through sialic acid to promote ovarian cancer tumor growth and progression. Oncogene. 2017;36(28):4014-24. Epub 20170313. doi: 10.1038/onc.2017.37. PubMed PMID: 28288142; PMCID: PMC5509519.
12. Rawat V, Malvi P, Della Manna D, Yang ES, Bugide S, Zhang X, Gupta R, Wajapeyee N. PSPH promotes melanoma growth and metastasis by metabolic deregulation-mediated transcriptional activation of NR4A1. Oncogene. 2021;40(13):2448-62. Epub 20210305. doi: 10.1038/s41388-021-01683-y. PubMed PMID: 33674745; PMCID: PMC8026604.
13. Nagarajan A, Dogra SK, Sun L, Gandotra N, Ho T, Cai G, Cline G, Kumar P, Cowles RA, Wajapeyee N. Paraoxonase 2 Facilitates Pancreatic Cancer Growth and Metastasis by Stimulating GLUT1-Mediated Glucose Transport. Mol Cell. 2017;67(4):685-701 e6. Epub 20170810. doi: 10.1016/j.molcel.2017.07.014. PubMed PMID: 28803777; PMCID: PMC5567863.
14. Pan L, Hong C, Chan LN, Xiao G, Malvi P, Robinson ME, Geng H, Reddy ST, Lee J, Khairnar V, Cosgun KN, Xu L, Kume K, Sadras T, Wang S, Wajapeyee N, Muschen M. PON2 subverts metabolic gatekeeper functions in B cells to promote leukemogenesis. Proc Natl Acad Sci U S A. 2021;118(7). doi: 10.1073/pnas.2016553118. PubMed PMID: 33531346; PMCID: PMC7896313.
15. Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U S A. 2018;115(15):E3509-E18. Epub 20180326. doi: 10.1073/pnas.1802691115. PubMed PMID: 29581297; PMCID: PMC5899497.
16. Bugide S, Gupta R, Green MR, Wajapeyee N. EZH2 inhibits NK cell-mediated antitumor immunity by suppressing CXCL10 expression in an HDAC10-dependent manner. Proc Natl Acad Sci U S A. 2021;118(30). doi: 10.1073/pnas.2102718118. PubMed PMID: 34301901; PMCID: PMC8325240.
17. Chibaya L, Murphy KC, DeMarco KD, Gopalan S, Liu H, Parikh CN, Lopez-Diaz Y, Faulkner M, Li J, Morris JPt, Ho YJ, Chana SK, Simon J, Luan W, Kulick A, de Stanchina E, Simin K, Zhu LJ, Fazzio TG, Lowe SW, Ruscetti M. EZH2 inhibition remodels the inflammatory senescence-associated secretory phenotype to potentiate pancreatic cancer immune surveillance. Nat Cancer. 2023;4(6):872-92. Epub 20230504. doi: 10.1038/s43018-023-00553-8. PubMed PMID: 37142692; PMCID: PMC10516132.
18. Zhang Z, Luo L, Xing C, Chen Y, Xu P, Li M, Zeng L, Li C, Ghosh S, Della Manna D, Townes T, Britt WJ, Wajapeyee N, Sleckman BP, Chong Z, Leavenworth JW, Yang ES. RNF2 ablation reprograms the tumor-immune microenvironment and stimulates durable NK and CD4(+) T-cell-dependent antitumor immunity. Nat Cancer. 2021;2(10):1018-38. Epub 20211022. doi: 10.1038/s43018-021-00263-z. PubMed PMID: 35121884; PMCID: PMC8809507.
Honors
- Research Scholar Grant, American Cancer Society, 2016
- Yale Cancer Center Translational Cancer Research Excellence Prize, Yale Cancer Center, Yale University, 2014
- IASLC Young Investigator Award, International Association for the Study of Lung Cancer, 2013
- Young Investigator Award Melanoma Research Alliance, 2013
- Kimmel Scholar for Translational Cancer Research. Sidney Kimmel Foundation for Cancer Research, 2012
- Hollis Brownstein Award for Leukemia Research, Leukemia Research Foundation, 2011
- AACR Centennial Cancer Research Award for Childhood Cancer Research American Association for Cancer Research, 2010
Education
Graduate School
PhD: Indian Institute of Science
Postdoctoral Training: University of Massachusettes Medical School
Contact
Office
Kaul Human Genetics Building
Room 540A
720 20th Street South
Birmingham, AL 35294-0024
Phone
(205) 934-5331
Email
nwajapey@uab.edu