 These findings in zebrafish and human heart muscle cells have clinical implications for patients with atrial fibrillation.Millions of adults have atrial fibrillation — an irregular beating of the upper chambers of the heart that yields increased risk of heart failure, stroke and death. Many genetic mutations in the developing fetus can lead to adult atrial fibrillation, including mutations that shorten the massive protein titin in cardiac muscle cells.
These findings in zebrafish and human heart muscle cells have clinical implications for patients with atrial fibrillation.Millions of adults have atrial fibrillation — an irregular beating of the upper chambers of the heart that yields increased risk of heart failure, stroke and death. Many genetic mutations in the developing fetus can lead to adult atrial fibrillation, including mutations that shorten the massive protein titin in cardiac muscle cells.
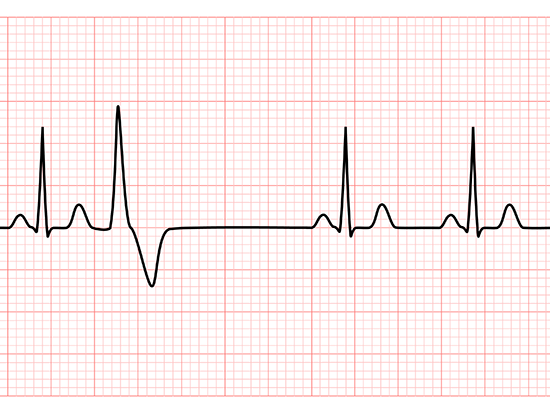
Now, in a study in zebrafish and human heart muscle cells, researchers show that a tiny deletion in the A-band of titin — the loss of just nine amino acids out of more than 27,000 to 35,000 amino acids of an intact titin protein — causes a developmental defect in the embryonic heart that leads to adult arrhythmia.
Researchers at the University of Alabama at Birmingham Marnix E. Heersink School of Medicine and at the University of Illinois Chicago found that the small internal deletion in titin that led to developmental abnormalities caused unexpected ion channel remodeling in heart muscle cells, creating an increased potassium ion current called /ks (pronounced “eye k s”).
Strikingly, researchers found that pharmacologically blocking the altered /ks current improved atrial contractility in the nine amino acid-deletion zebrafish embryos, and it similarly improved contractility and prevented atrial fibrillation in the nine amino acid-deletion human cells.
“This work has potential clinical implications for both pediatric and adult patients,” said Ankur Saxena, Ph.D., a UAB associate professor in the Department of Cell, Developmental and Integrative Biology. “With titin serving as an electromechanical bridge between sarcomeric structures and ion channels, developing drug treatments targeting ion channel remodeling may restore and maintain sinus rhythm and improve contractility, as well as improve the long-term outcome for patients.”
Saxena and Dawood Darbar, M.D., of the University of Illinois Chicago, are corresponding authors of the study, published in the journal iScience.
In studying the effects of the nine-amino acid deletion in zebrafish embryos and adults as well as human induced pluripotent stem cell-derived atrial cardiomyocytes, the UAB and University of Illinois Chicago researchers discovered that zebrafish embryos homozygous for the nine-amino acid deletion showed a transient reduction in ventricular function, with smaller size, reduced contraction and slower blood circulation; however, the ventricle recovered within a few days. In contrast, embryonic and adult mutant zebrafish had persistent atrial enlargement, and adult mutant zebrafish had atrial fibrillation. In both the adult zebrafish atria and human atrial cardiomyocytes, mutated titin yielded sarcomeric disorganization and reduced contraction.
 Ankur Saxena, Ph.D.The researchers next explored how transient ventricular and persistent atrial embryonic defects mechanistically led to adult atrial fibrillation. Previous work had shown that the hormone atrial natriuretic peptide, or ANP, is overexpressed in response to ventricular dysfunction via an increased /ks, known as the slow delayed rectifier potassium current. In agreement with that, co-first authors of the study, Xinghang Jiang, a postdoctoral fellow in the UAB Department of Cell, Developmental, and Integrative Biology, and Olivia T. Ly, University of Illinois Chicago, and colleagues found aberrant ANP expression and changes in the expression of proteins that form the potassium channel in both the mutant zebrafish atria and human cardiomyocytes. Furthermore, knockdown of ANP improved atrial contraction, and voltage clamp experiments showed potassium channel remodeling, with significantly higher peak /ks density.
Ankur Saxena, Ph.D.The researchers next explored how transient ventricular and persistent atrial embryonic defects mechanistically led to adult atrial fibrillation. Previous work had shown that the hormone atrial natriuretic peptide, or ANP, is overexpressed in response to ventricular dysfunction via an increased /ks, known as the slow delayed rectifier potassium current. In agreement with that, co-first authors of the study, Xinghang Jiang, a postdoctoral fellow in the UAB Department of Cell, Developmental, and Integrative Biology, and Olivia T. Ly, University of Illinois Chicago, and colleagues found aberrant ANP expression and changes in the expression of proteins that form the potassium channel in both the mutant zebrafish atria and human cardiomyocytes. Furthermore, knockdown of ANP improved atrial contraction, and voltage clamp experiments showed potassium channel remodeling, with significantly higher peak /ks density.
“Elucidating the molecular mechanisms by which developmental defects lead to increased risk of atrial fibrillation in adults has been challenging, with rare known examples,” the researchers wrote in the study. “Here, to explore the relationship between developmental defects and the pathogenesis of atrial fibrillation, we deleted just nine amino acids in titin’s A-band and identified an unexpected role for this giant protein in mediating both ion channel-dependent remodeling and impaired atrial contractility. Notably, early cardiac dysfunction and recovery lead to aberrant ANP expression and ion channel remodeling, with potential implications for how subtle structural mutations might cause subclinical abnormalities that lead to atrial fibrillation in adulthood.”
Co-authors in the study, “Transient titin-dependent ventricular defects during development lead to adult atrial arrhythmia and impaired contractility,” along with Jiang, Ly, Saxena and Darbar, are Hanna Chen, Ziwei Zhang, Beatriz A. Ibarra, Mahmud A. Pavel, Grace E. Brown, Arvind Sridhar, David Tofovic, Abigail Swick, Richard Marszalek, Fritz Navales, Salman R. Khetani, Jalees Rehman and Yu Gao, University of Illinois Chicago; and Carlos G. Vanoye and Alfred L. George Jr., Northwestern University, Chicago, Illinois.
Support came from National Institutes of Health grants HL138737, HL150586, HL148444, GM133416 and HL139439; and the UAB Heersink School of Medicine. At UAB, Cell, Developmental, and Integrative Biology is a rapidly growing basic science department in the Heersink School of Medicine.
Saxena moved to UAB from the University of Illinois Chicago last fall.