


Cystic fibrosis (CF) can feel like suffocation in slow motion. Mutations in the CFTR gene, a key regulator of hydration status, cause sticky mucus to clog the airways of people with the disease.
It can take hours of physiotherapy each day to ease labored breathing, and the mucus is a haven for bacteria that bring a constant threat of potentially fatal infections.
 Steven Rowe
Steven RoweSteven Rowe, M.D., first encountered this suffering as a teenage counselor at a camp for children with medical needs in the early 1990s. “Even if they did everything right, the disease would just get worse,” he recalls. “The clock was always ticking.” Rowe parlayed that experience into a mission for his career: Discover what goes wrong in CF and find new ways to treat it.
At the time, physicians had few options for patients. Everything changed in 2012 when the Food and Drug Administration approved ivacaftor, a drug that can fix the faulty proteins at the heart of CF. Patients in clinical trials suddenly were clearing mucus and making major gains in lung function and quality of life.
“It’s one of the most remarkable stories in modern medicine,” says Rowe, the Nancy R. and Eugene C. Gwaltney Family Endowed Chair in Medical Research in the School of Medicine. UAB has played a central role in that story through the Gregory Fleming James Cystic Fibrosis Research Center, which Rowe now directs. Decades of basic-science discoveries in the center, named for Alabama governor Fob James’s second son, who died of CF at age 8, paved the way for Rowe’s lab to refine tests crucial to evaluating the compounds that led to ivacaftor. His contributions to clinical trial design, along with UAB’s reputation, have made Birmingham a hub for testing new treatments, giving Alabama patients an early chance to benefit from their success.
Right now, about 15 percent of the 30,000 Americans with CF have highly effective therapies available, says Rowe. But research led by UAB scientists is on the verge of raising that percentage far higher—and their CF discoveries may also benefit millions of patients with other diseases, from COPD to asthma.

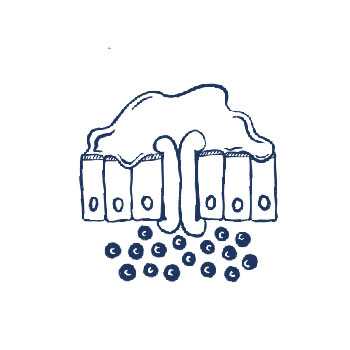
CF up close: In people without CF (above), CFTR regulates channels that allow chloride and water to pass through cell walls and hydrate airways. In people with CF (below), CFTR mutations can prevent these channels from opening properly or cause other malfunctions, leading to the formation of thick mucus that coats the airways.
Super friends
Like comic book heroes, the new CF drugs seem to work best when they join forces: “Potentiators,” like ivacaftor, force CFTR protein channels to stay open, and “corrector” drugs help mutated proteins reach the cell surface. In 2017, Rowe and his colleagues led national studies showing that a combination of ivacaftor and a corrector known as tezacaftor substantially improved lung function among patients who have two mutations: F508del, which is the most common CF mutation, plus a relatively rare “residual function” mutation. Around 10 percent of people with CF have both mutations. Last October, the team announced an even bigger breakthrough. A UAB-led trial found that ivacaftor and tezacaftor, combined with a new corrector, VX-659, provided a one-two-three punch with the potential to improve lung function for 90 percent of all CF patients—those with either one or two copies of the F508del mutation.
Rowe and his colleagues are beyond excited by the results—and the possibilities. Large-scale studies of the triple therapy and another similar drug combo are under way, which means “we’re on the brink of seeing this through from vision to execution and availability,” Rowe says. With further development, the treatment could help push average life expectancy for people with CF—currently around 47 years of age—even higher, he adds.
 David Bedwell
David BedwellMaking sense of nonsense
Severe forms of CF often are triggered by “nonsense” mutations, which are essentially premature “stop” codes in the CFTR gene. They prompt the cell to build incomplete proteins that are quickly destroyed. In 1996, David Bedwell, Ph.D., and colleagues reported that some antibiotics could encourage the cell to ignore, or “readthrough,” the stop signs and make complete proteins.
It was a revolutionary idea then, says Bedwell, associate director of the UAB CF Center and chair of the Department of Biochemistry and Molecular Genetics. But today, researchers estimate that up to 11 percent of all mutations that cause genetic diseases are the result of nonsense mutations, Bedwell explains. “These drugs could help millions worldwide.”
Already, readthrough drugs are used to treat nonsense-caused Duchenne’s muscular dystrophy in Europe, and Bedwell has seen promising results from them in addressing Hurler syndrome. Now he and partners at Southern Research and the Cystic Fibrosis Foundation are searching for a powerful readthrough drug for CF. So far, they have sifted through 750,000 compounds to identify about 150 promising candidates.
 George Solomon
George SolomonThe nose knows
Of the 2,000 known mutations in CF, only a handful are widely studied. Many patients have mutations so rare that it’s difficult to know which drugs may help, which can lead to expensive choices when the cost of some medications can exceed $300,000 a year. “These are the ‘lost patients’ in CF,” says CF Center scientist George Solomon, M.D. To address their needs, he and fellow CF Center scientist Jennifer Guimbellot, M.D., Ph.D., are taking a precision medicine approach, working to create individualized tests from patients’ own cells. A small sample from the lining inside a patient’s nose enables Guimbellot to make thousands of “nasospheroids”—rings of cells the width of a human hair. These rings may respond to CF drugs in the same way the whole patient would, she has found. “Our hope is that simple lab tests using nasopheroids could tell doctors, within a few days, whether a patient with a rare mutation will benefit from a particular drug,” Guimbellot says.
The same technique eventually could help tailor treatments for all CF patients, even those with more familiar mutations. “You can see 20 patients with the exact same [CFTR] mutation, and they have different responses to the same drug,” Guimbellot says. “I envision a time when everyone will get a personalized profile, instead of relying on trial and error.”
 Jennifer Guimbellot
Jennifer GuimbellotSmoking out a killer
Once UAB researchers understood ivacaftor’s mucus-clearing powers, they sought out other mucus-associated conditions that could benefit from the treatment. That led them to chronic bronchitis. Caused largely by smoking, the disease affects millions. It, in turn, is a major cause of chronic obstructive pulmonary disease (COPD), one of the world’s leading causes of death.
Tests in Rowe’s lab showed that cigarette smoke impairs CFTR function. Working with Mark Dransfield, M.D., medical director of UAB’s Lung Health Center, Rowe and colleagues are now testing ivacaftor in patients with chronic bronchitis. Early results have shown some “pretty dramatic responses,” Dransfield says.
Like CF a decade ago, chronic bronchitis has no therapies that get to the root of the problem, Dransfield notes. Success with ivacaftor could make a big impact, particularly in Alabama, which ranks second in the nation in the prevalence of chronic bronchitis. “We’re hoping to apply a drug developed for a few thousand people with CF to up to 10 million people with chronic bronchitis,” Dransfield says.
 Mark Dransfield
Mark DransfieldVision quest
Understanding the interplay among mucus, the cells lining the airway, and cilia (the hairlike structures that push mucus through the airway), is crucial to further breakthroughs in CF and other mucus-related diseases, including COPD and asthma. To get a closer look, UAB researchers and partners at Harvard Medical School developed micro-optical coherence tomography (microOCT). The advanced imaging technology is “basically ultrasound with a laser,” Rowe explains. “It lets us see things no one has ever seen before—features we didn’t imagine,” including elements of the malfunctioning biology that could be targets for future treatments. The team now has created a miniaturized version of microOCT for clinical use, which could soon offer doctors a real-time look at individual patient responses to therapy.
Seed money from the Gwaltney Endowed Chair was crucial to getting the microOCT project—and many other bold breakthroughs from UAB’s CF Center—off the ground, Rowe notes. “With that support, we were able to take this shot,” Rowe says. “That’s always been our philosophy. The more shots we can take, the more we can make a difference in the lives of our patients.”
 These watercolor illustrations (above and at top), created by Sophie McVicar, a senior UAB chemistry and art major from Chico, California, depict the 3-D structure of CFTR, the protein responsible for moving chloride ions out of cells. Mutations in this protein cause cystic fibrosis. The ribbon model of CFTR visualizes the ways in which the protein interacts with itself.
These watercolor illustrations (above and at top), created by Sophie McVicar, a senior UAB chemistry and art major from Chico, California, depict the 3-D structure of CFTR, the protein responsible for moving chloride ions out of cells. Mutations in this protein cause cystic fibrosis. The ribbon model of CFTR visualizes the ways in which the protein interacts with itself.